医院新闻



近日,在中组部“组团式”援滇医疗队(简称“医疗队”)专家及复旦大学附属儿科医院(简称“儿科医院”)专家组的共同努力下,金平县人民医院成功诊断了金平县域内首例脊髓性肌萎缩(SMA)患儿。明确诊断将为患儿的治疗提供了重要技术支持,更为金平及周边地区脊髓性肌萎缩患儿带来了希望。标志着金平县人民医院在罕见病、疑难病的诊疗能力步入一个新的台阶。
5岁的患儿,因行走姿势异常来到金平县人民医院就诊,此前曾到省、州级多家医院就诊,诊断为“肌营养不良”、“先天性髋臼发育不良”,外院准备为其行截骨手术。
在听闻医疗队进驻金平县人民医院的消息后,家长专程带着孩子前来咨询。医疗队专家张赟健(现金平县人民医院儿科主任)和专家陈增淦(现金平县人民医院骨创伤外科主任)对该患儿进行了详细的病史询问和仔细的体格检查,发现该患儿行走明显鸭步,查体肌张力降低,腱反射减弱,Gower征阳性,高度怀疑其为神经肌病,目前不应该进行截骨手术治疗。
张赟健主任为患儿做了肌酶检查,排除了常见的进行性肌营养不良,考虑脊髓性肌萎缩(SMA)或先天性肌病可能。为明确患儿病因,张赟健主任与大后方儿科医院神经肌病李西华主任、胡超平主治医师沟通讨论后决定,先通过儿科医院分子医学中心为患儿进行脊髓性肌萎缩基因检测。经过基因检查,患儿确诊为SMA。该病例是在沪滇携手下确诊的首例SMA患儿。该病例的诊断也提高了当地医务人员对SMA的认识。
患儿的情况牵动了医院同仁的心,金平县人民医院党委书记龙进勇,医疗队队长唐文斌(现金平县人民医院院长)、副院长缪应生、儿科主任周文高度重视,第一时间为患儿开辟绿色通道,为开展及时救治提供条件。
张赟健主任再次积极与大后方复旦大学附属儿科医院协调,邀请儿科医院SMA-MDT团队进行远程会诊,希望能够对患儿后续的治疗方案、疾病的长程管理及并发症的防治等方面进行专业的指导。




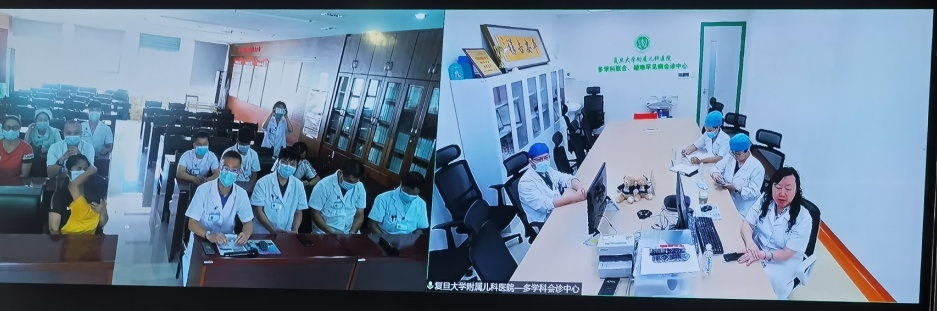
中华医学会儿科学分会罕见病学组组长王艺教授、神经科李文辉副主任医师、朱小妹主治医师、骨科钱闯主治医师、康复科李惠主管治疗师、营养科龚晓妍主管营养师等多位知名专家出席了会诊。经过专家会诊,结合患儿临床表现及儿科医院基因检查结果,患儿3型SMA诊断明确。在原发病治疗方面,专家讨论后建议予疾病修正药物诺西那生钠鞘内注射靶向治疗。专家们对患儿目前神经肌肉及骨骼系统功能进行详细评估,为患儿制定了个体化康复治疗方案,并在线指导家长,包括对患儿开展训练、营养指导、家庭设施改善,并为患儿家庭再生育需求提供遗传咨询。
此次儿科医院与县人民医院SMA-MDT远程会诊的开展,再次让金平患儿在县域内就能得到儿科医院全国顶级专家的诊疗服务,为边疆人民的就医提供了更多的便利。
科普知识
一、什么是脊髓性肌萎缩
脊髓性肌萎缩(SMA)是一种罕见的遗传性神经肌肉病,其发病率为1/10000-1/6000,为常染色体隐性遗传病,由运动神经元存活基因1(SMN1)突变所致,导致肌无力和肌萎缩。随着病情进展,肌无力可进一步导致骨骼系统、呼吸系统、营养不良等多系统功能异常,可致死致残,其中呼吸衰竭是最常见的死亡原因。
二、SMA有哪些临床表现
患儿临床表现差异性大,发病年龄可以从出生前(宫内发病)开始,表现为胎动减少,也可以在成年后。根据发病年龄、病情进展速度,可将SMA分为4型:
SMA Ⅰ型:1/3在宫内表现为胎动减少,出生时为松软儿,平均发病年龄在生后1个月。表现为全身松软无力,严重肌张力低下,大多数患儿出现吸吮和吞咽困难,呼吸困难,不会抬头或翻身,没有独坐能力。此型患儿肌无力进行性加重,最终失去所有自主运动能力,多数于2岁内死于呼吸衰竭。
SMA Ⅱ型:患儿生后6个月内发育正常,通常在18个月内出现症状,患儿不能独立行走,随着病程进展,出现吞咽困难、脊柱侧弯、呼吸功能不全。一般可存活至10~20岁,智力正常。
SMA Ⅲ型:生后1年内运动发育正常。从幼儿期至青少年期均可发病,可以获得独立行走的能力。3岁前起病者50%在14岁左右失去独走的能力,后期出现脊柱变形,可以存活至中年,智力正常。
SMA Ⅳ型:又称成人型SMA。多在30~60岁发病,出现肢体近端无力,病情进展缓慢。
三、SMA如何诊断
SMA通常通过临床表现可做出诊断,分子遗传学检测SMN1基因可确诊。
四、SMA如何治疗
2019年2月,全球首个治疗SMA的疾病修正治疗药物诺西那生钠注射液在中国获批上市,同年10月正式应用于临床,开启了国内SMA精准治疗的新时代。诺西那生钠是一种反义寡核苷酸药物,通过鞘内注射给药用于治疗各型SMA患者。SMA的标准化管理需神经、呼吸、骨科、营养、消化、康复等多学科医生共同参与。
专家简介








服务号

订阅号

